When we fill porous materials with dye molecules of the right size, we obtain useful compounds for solar energy technology. These compounds can transfer solar energy efficiently because pores and channels fit to the dyes “like a glove”. In this way, molecules are forced to stay in line, and energy can easily pass from a molecule to the next one in the line. If we knew in detail the structure of the dye arrays, we’d have better chances to improve these compounds.
Unfortunately, the precise positioning of the molecules inside the pores is very hard to determine. Recently, we solved this problem for a class of particularly efficient dyes filling the channels of zeolite L. Key to success was diversity within the team, which favored the combination of multiple techniques involving both experiments and calculations.
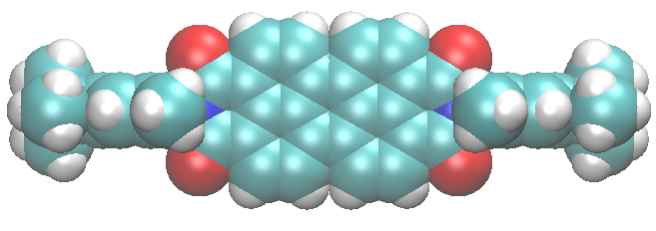
The useful properties of these materials arise from the arrangement of dye molecules inside the porous host, which depends on the interactions among molecules and with the porous host. After this work, now it seems we understand a little better these complex materials. Indeed, our dyes are linear, symmetric and fit to the zeolite channels. Yet they adopt a slightly asymmetric positioning to maximize the interactions with the zeolite cations, which stabilize the compound.
Perylene-bisimide dye (cyan) in zeolite L (gray). The purple spheres represent the zeolite potassium cations
This work also suggests some possible ideas to improve these compounds by modifying either the porous container (the “host”) or the dye molecule (the “guest”). In my view, this is also a good example of how computational modeling may help to rationalize experimental results in apparent contrast with each other, yielding a consistent picture of a useful and intriguing material.
Gigli et al. (2018) “Structure and Host–Guest Interactions of Perylene–Diimide Dyes in Zeolite L Nanochannels”J. Phys. Chem. C 122, 6, 3401-3418
TheTwitter Poster Conference is an annual event organized by the Royal Society of Chemistry, which consists of sharing chemical research using tweets. You may take part either by tweeting an image of your poster or by commenting on other poster (doing both is better, in my view). As in a traditional conference, you see great research, are asked interesting questions, meet old friends, and come in contact with new colleagues or potential collaborators. Only, at the twitter conference this happens 24 hours non-stop on global scale; so, it’s a good idea to get there prepared!
I enjoyed so much my 2017 participation that I couldn’t miss the 2018 edition. Of course, it was awesome, and I am grateful to the organizers, the sessions’ chairs, and all participants, particularly those with whom I interacted. Indeed, I have learnt new stuff, seen exciting science, been inspired, without moving from my office and paying any conference fees. Really cannot ask for more. Many thanks to all of you!
Tweeting posters is not trivial. For optimal readability, you should keep into account, for example, that mobile phones have small screens, and that Twitter images are resized and cropped down – so, it would be better to prepare the poster in landscape format. These and other useful tips can be found in this excellent post. I came across it when the event was over, but it would surely be useful in the future.
Below you can find my poster, illustrating the fruitful collaboration between calculations and diffraction experiments at high-pressure conditions.
Besides water and ethanol in ferrierite (discussed in this post), the poster shows our new work on a host-guest compound of zeolite L and fluorenone dye under high pressure. These host-guest materials have excellent optical properties, useful for many applications, from solar cells to sensing in medical technology. Knowing their structure and working principles could help improve their performances – that’s why we try so hard to understand dye-zeolite composites at molecular level.
Basically fluorenone inside the channels of zeolite L forms a molecular ladder, which is very stable at room conditions because the carbonyl groups of the dye interact very strongly with the potassium cations of the zeolite.
Is this peculiar structure also stable under GPa pressures?
According to experiments and simulations, the answer is apparently yes! Our composite maintains its structure, and the interactions between the dye and the zeolite cations become stronger. The exceptional resilience of this material to compression highlights its outstanding mechanical properties. These are important to extend the application of dye-zeolite composites beyond room-pressure conditions.
Our work on ethanol and water in ferrierite, published here and blogged in my previous post, has been recently covered by MRS Bulletin in an excellent news article – “High pressure and small spaces create order from disorder” by science writer Tim Palucka. Some time ago, I had a very pleasant communication with Tim about the main ideas and results of the paper. That interview also helped me a lot to understand how science communication is done professionally. The piece by Tim really does a great job in explaining the scientific background, the main findings and the perspectives of our research – and, of course, all of us are so happy about it!
MRS Bulletin contains other interesting news articles, which are very useful to get a first impression about what’s going on in the many diverse areas of materials science – we’re very proud to be featured there! Big thanks, therefore, to MRS Bulletin and Dr. Palucka for the awesome coverage, and to Prof. Gion Calzaferri for commenting on our work as an external expert. A pdf version of the news article is freely available at MRS Bulletin (Volume 42, Issue 3, pp. 176-177, DOI: https://doi.org/10.1557/mrs.2017.38 ), while the illustration showing the arrangement of water and ethanol in the zeolite is just here below:
What happens to a liquid mixture when it is driven by pressure into an initially empty container? What if the container has an ordered pattern of molecular-sized pores? To answer this question, we prepared a sort of good vodka drink – three parts water, and one ethanol – and we injected it into the pores of an hydrophobic container – the zeolite ferrierite. As hydrophobic materials don’t like water and don’t care about drinks, we had to be very drastic: we used a diamond anvil cell. In this apparatus, the sample – the empty container and the mixture, in our case – is compressed between the tips of two opposing diamonds and experiences huge pressures – about 10.000 times the normal atmospheric pressure. At these conditions, matter is subjected to unimaginable forces, comparable to internal atomic forces: this means that strange, unexpected phenomena could show up. Now, let’s combine the power of high-pressures with the ordering effect of the pore matrix and see what happens to our mixture.
Just to start with, the water-ethanol mixture – the pressure-transmitting medium – enters the pores of the matrix. But how do the molecules occupy the pores? You don’t need to be a chemist to know how it is difficult to separate alcohol from water. This is a critical issue also for sustainable processes – such as the production of biofuels.
Thanks to high pressure and to the porous matrix – and with the help of computational modeling – here we obtained the separation of ethanol from water, and the formation of a beautiful pattern of clusters. The clusters – rows of ethanol dimers, and square water tetramers – occupy different regions of the host matrix and alternate like tiles forming a nice molecular mosaic – a “two-dimensional architecture” – inside the porous host. What’s really exciting about it is that the ordered pattern, created by high pressure, also remained stable by bringing the material back to atmospheric pressure. This means that using high pressures and porous hosts, we can create new materials, which are stable at normal conditions, and could potentially be exploited in applications.
The metamorphosis of the initial water-ethanol solution into a beautiful two-dimensional pattern remains somewhat mysterious. More in general, how organization arises from chaos is still one big question in science. However, our molecular dynamics simulations show that water molecules, already inside the pores, can spontaneously self-organize in square tetramers:
The final result, is the formation of the stable two-dimensional architecture of water and ethanol clusters. As the movie shows, the molecules move, but the clusters do not break apart. – even upon returning to room pressure.
Perspectives
Disclosing the way in which molecules and nanoparticles assemble at high pressure conditions, under the guidance of a suitable matrix would be a great and intriguing challenge for future studies. Another one would be the actual production of technologically relevant materials through the combined use of pressures and suitable porous matrices. These goals could be achieved only through a close collaboration between experiment and theory – a synergy which has been at the very origin of the present work.
In a wider perspective, understanding the behavior of matter at high pressures is of central relevance in science, as explained in this excellent introductory feature article. Pressure effects are ubiquitous, in chemistry, physics, earth and planetary sciences, as well as in many industrial processes and technological applications. High-pressure conditions are also hypothesized to explain the origin of complex chemistry and life. The study of this exotic regime, so different from our everyday-life, may reveal plenty of phenomena which would be hard to imagine based on our experience.
Reference: Irreversible Conversion of a Water–Ethanol Solution into an Organized Two-Dimensional Network of Alternating Supramolecular Units in a Hydrophobic Zeolite under Pressure, by Rossella Arletti, Ettore Fois, Lara Gigli, Giovanna Vezzalini, Simona Quartieri, and myself. Angewandte Chemie 2017 – DOI: http://dx.doi.org/10.1002/anie.201610949 http://dx.doi.org/10.1002/ange.201610949
Special thanks to Andrea Stangoni (@andrea_stangoni), author of the cover artwork. His image summarizes the ideas of our work much more beautifully than my blog post!
How can a snake swallow a mouse bigger than its mouth?
Weird as it seems, questions like this emerge very often at the molecular scale. For example, we can fill porous materials with molecules larger than the diameter of the pores: in this way, we may obtain devices for energy and health applications. What makes this useful process possible? Flexibility is the key: both the porous host (the “snake”) and the molecule (the “mouse”) must deform for the process to occur. But here, contrary to the mouse-snake case, cooperation between the two partners is needed.
We captured the passage of a bulky molecule through the very narrow opening of one of these pores. We did this by computer simulations, because it is very hard to get such information experimentally. To get an idea of what we found, you don’t even need to read the paper – and i’m not kidding. Just look at the movie below!
What we’ve seen first, is that the pore is slightly larger at its entrance. This surely helps the molecule to go in.
Second: contrary to the mouse, which would escape the snake as fast as it could, the molecule is indeed “magically” drawn to the pore entrance – by electrostatic forces.
“So what?” – you may say.
Keep in mind that the molecule is still larger than the pore opening. No kind of “fatal attraction” could do the trick, in a world of rigid bodies.
We’ve found that the molecule can pass through the opening and slip inside the pore only because it’s flexible, and its motion is “in tune” with the vibrations of the porous matrix. All this factors cope to make the entrance process more favorable than the exit process – that’s why the molecule gets finally swallowed by the pore, and remains trapped inside the material.
For me, it was very nice to see how bulky molecules manage to pass through narrow openings and travel inside a porous material. But finding out the reason why they stay inside was, probably, even more exciting: because it explains how materials of this kind can form and remain stable. Which is exactly one of the things you may need, in the quest of easier and smarter ways to produce better materials.
As we have to give credit where credit is due, i must confess that i borrowed the mouse-and-snake idea used in this post. But you’ll never know from whom. Me neither: (s)he was an anonymous referee of the paper. I am very grateful to this person: i can hardly imagine a nicest way to sketch our work.
Update:
Many thanks, of course, also to ChemComm for the cover!
Teaching is an important part of my daily activity. It is time-consuming, requires a lot of energy and involves an emotionally intensive effort so, why not blogging about it?
Explaining physical chemistry to undergraduate students is by no means an easy task. Many students consider it too difficult -which is bad – and boring – which is even worse. When last September i was asked to set up a new physical chemistry course from scratch, i went into panic mode. Mission impossible. How could i make this course interesting to students aspiring to become organic or analytical chemists?
Problem n. 1: The content. There were already several courses on theoretical chemistry. I wanted something different: with solid content, but new and appealing. Two important topics were not sufficiently covered by other courses: electrostatics/electric currents, and intermolecular forces – quite useless and unexciting things, to students’ eyes. My idea was to show them that knowledge of those boring topics could unlock the door to molecular electronics and supramolecular chemistry. To make students willing to learn that stuff – quite an ambitious goal – I used examples, borrowed from the recent literature and even from my own research work.
Problem n. 2: The name. Names of physical chemistry courses often appear obscure and discouraging, to students. Fortunately I shared my worries with some colleagues, and, after some brainstorming, we converged on “applied physical chemistry: from molecules to devices”. Don’t know if this was a good choice – only time will tell. But all students of the master degree chose to follow it … because of the “applied”, i suspect. Anyway, that was a good start, at least.
Problem n. 3: The lab. That is, the practical part of the course. Why not a compchem lab? Despite being a computational chemist i never had the occasion of doing that before – i had to teach other courses – and again i felt overwhelmed. Many of the students had never seen any of the most basic Linux commands, not even used a quantum chemistry code. Once again i shared my thoughts with colleagues, and looked through the web in search of suggestions. There’s a lot of excellent material, but, unfortunately, often too advanced for the needs of my students – most of them at their very first exposure to computational chemistry.
My solution was to schedule three 4-hour sessions. Very schematically, the objectives were the following:
1) Learn the basic Linux commands, prepare Gaussian-09 input files and run energy calculations for simple systems -a water molecule, a water molecule dimer, ethylene and benzene. Plot the electron density and the electrostatic potential surfaces and deduce from them the possible types of intermolecular interactions.
2) calculate by points the potential energy curve as a function of distance for a sodium cation interacting with a benzene molecule. Plot the curve and compare the resulting energy minimum distance with that obtained from a geometry optimization of the full system benzene-Na+.
3) (this was the most exciting part) – run energy calculations on components of molecular machines, e.g. small diazobenzene axles and crown ether rings, and try to discuss the possible intermolecular interactions between these components on the basis of the electrostatic potential maps. In the picture below, you can see the students actually doing such little exercise!
I’m not sure that this was the right way to go. Of course, everything can be done better -some important issues, on e.g., accuracy and basis set choice, were necessarily swept under the carpet, but I had the feeling that the students sort of enjoyed their first approach to modeling.
During this weekend i tackled a challenging task: to try to explain one of my recently published papers with an infographic. My first thought was to write a blog post (maybe i’ll do it as well), but i was intrigued by the idea of lumping a couple of years of work into a few tiny lines. Although attracted by the immediacy of infographics, i never used this tool, and it sounded just the right moment to give it a go. So, i went to the Canvas site and chose a fitness club advertisement as a template. After a bit of playin’ around, that’s what i’ve got:
I admit i’m quite happy with it, even if the making process was not plain sailing at all, at least for me. As a first-time user, i think that there’s room for improvement, and i’ll probably do some other attempts. Actually, i enjoyed creating this infographic!
I have published it (the infografic, i mean) in figshare (acceptance rate: 100%, publication fares: 0 €). That’s openaccess – free to download and use. Unfortunately, that’s not true for the paper – not enough funds to make it openaccess as well. Anyway, if you might want to give it a look, here’s the link:
Deiana, C., Fois, E., Martra, G., Narbey, S., Pellegrino, F. and Tabacchi, G. (2016), On the Simple Complexity of Carbon Monoxide on Oxide Surfaces: Facet-Specific Donation and Backdonation Effects Revealed on TiO2 Anatase Nanoparticles. ChemPhysChem. doi:10.1002/cphc.201600284
Another short explanation can be found here, with links to additional material.
To understand how a motor works you have to know how it is put together and how it can be disassembled. This is true at the molecular level as well. Amazingly complex molecular motors and machines are fabricated everyday, but how do they break down into their constituent pieces? Attracted by this question, we modeled two such species – called ‘rotaxanes’ – and made them break apart.
Typically, rotaxanes are made by two molecules: a ring-shaped one, the “wheel”, and an approximately linear one, the “axle”. What is great about them, is that you can modify the interactions between the ring and the axle by using an external signal. Which means that you can control the movements of these components through light, for example.
A ring (R)An axle (EE)Another axle (ZZ)
Using the three components shown above – one ring and two axles – you can build two different rotaxanes: R-EE, and R-ZZ. Both disassemble – or ‘dethread’ – when the axle exits from the ring. This process occurs differently for the two species: While the EE-one (video 1) dethreads in one single step:
the dethreading of the ZZ-axle (video 2) passes through an intermediate and a transition state:
Analyzing the two simulations, we identified the interactions between the molecular components which control the process at the molecular level. For example, we have seen that the elliptic shape of the ring opening is very important, as it can recognize the two different configurations of the terminal groups of the axles. This insight might help in the construction of light-powered molecular devices, of which these rotaxanes are important building blocks.
The results of this work are discussed in our paper (1), which is part of the ChemPhysChem special issue on molecular machines. You may get a nice picture of the field by taking a look at this issue. Its contributions well illustrate the concept that molecular machines -especially natural ones – are incredibly complex and fascinating systems, and we are just beginning to understand their inner workings.