Wearing sunscreen everyday decreases your risk of skin cancers and helps your skin maintain a healthier look. But how does a sunscreen work? The chemistry of a sunscreen lotion, nicely pictured in this infographics, is actually quite a complex matter. The key ingredients are molecules known as UV-filters, which can absorb UV light and then dissipate the energy harmlessly in the form of heat.
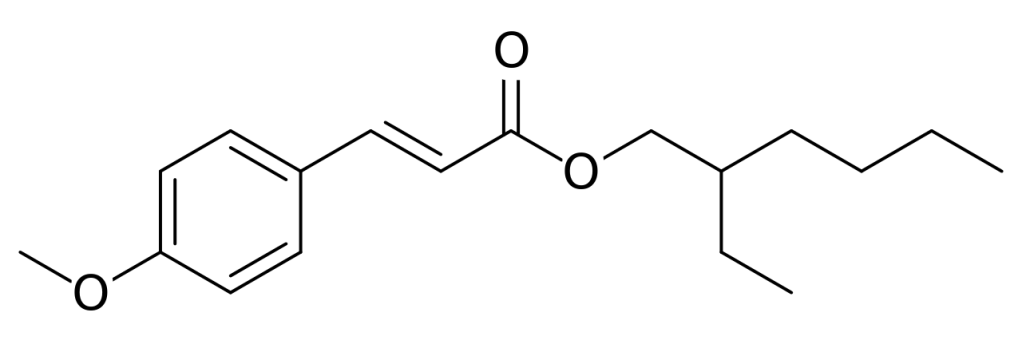
Octinoxate is one such molecules. The active part contains a ring system and conjugated double bonds. It has a flat structure – denoted as “trans“- and excellent UV-B filter properties. Unfortunately, its filtering action is degraded by light over time. Under sunlight, the molecule is converted to a less effective form, named “cis“. Understanding why this process occurs might help to improve the efficacy of sunscreen lotion and creams.
Chemical structure of trans-octinoxate (octyl methoxycinnamate). The left part of the molecule acts as UV-filter, while the right part – the alkyl chain – ensures optimal mixture in sunscreen lotions.https://commons.wikimedia.org/wiki/File:Octyl_methoxycinnamate.svg
We studied with accurate calculations the two forms of the octinoxate molecule. In contrast to what was previously thought, the less active cis form is slightly more stable than the commercially valuable trans form. This finding explains why the UV filter loses efficacy, and may suggest possible ways to limit the degradation processes.
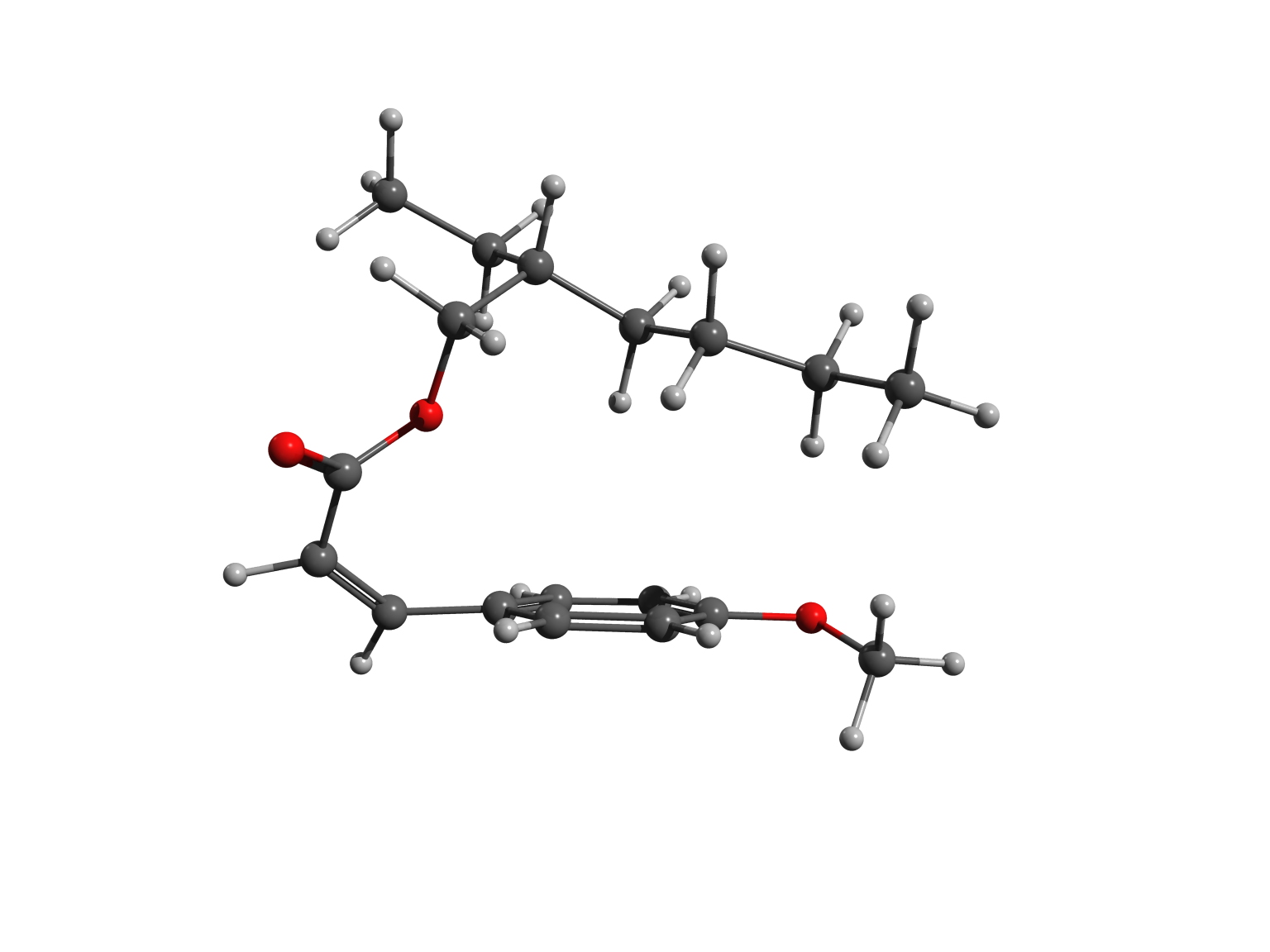
The two forms – or isomers – of the molecules have very different shapes. The trans is approximately flat, as shown in the left picture, while the cis is bent, as illustrated in the right image.
In cis octinoxate, the alkyl chain folds over the other part of the molecule, leading to a more compact shape, stabilized by dispersion interactions. Practically, the chain plays a key role, because it allows the filter to be easily dispersed in the lotions and creams we commonly use to protect our skin.
More technically, these subtle effects, highlighted by post-Hartree Fock calculations, are not fully captured by density functional approaches. All the tested functionals, including the B2PLYP double hybrid, predict the trans structure to be more stable than the cis one. This molecule is indeed a challenging system for any computational approach.
The striking difference of shape between nearly flat trans-octinoxate and folded cis-octinoxate suggests that this change of shape might help to dissipate the energy accumulated upon light absorption. Perhaps, this effect might also be beneficial to the sunscreen action. As a further step to understand the peculiarity of this molecule, the cis– and trans- forms of octinoxate could be also investigated via molecular dynamics simulations to better capture the differences in their complex behaviour.
The replacement of fossil fuel with sustainable alternatives free from environmental footprint is one of the most important challenges to combat climate change and meet the ever increasing energy demand of our planet. The sustainable production of hydrogen fuel through biomass-derived ethanol in Direct Ethanol Fuel Cells is a promising route, but the high costs and short lifecycle of platinum – which is still the preferred catalyst – are a serious problem. So, the quest to valid yet convenient substitutes to platinum is an open and challenging task.
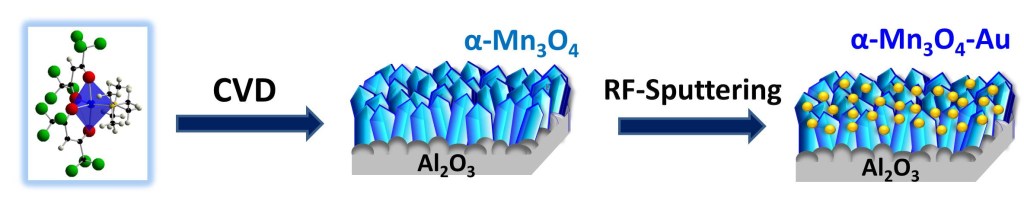
We (actually, my experimental colleagues) prepared an electrocatalysts for Ethanol Oxidation Reaction based on a low-cost and abundant metal oxide, namely manganese oxide. The fabrication strategy involves the growth of manganese oxide nanostructures on nickel foam scaffolds via plasma-assisted chemical vapor deposition and the functionalization with gold nanoparticles in low amount – as sketched in the picture below. That’s the magic of molecule-to-nanomaterials conversion!
The synthesized nanostructures have large surface area and show great performances as electrocatalysts in the ethanol oxidation reaction, comparing favourably with the best oxide-based catalysts known to date. We found that a very tiny amount of gold nanoparticles is sufficient to boost the catalytic activity of manganese oxide.
Our findings not only afford a convenient route for sustainable electrocatalysts, but also explain why our catalyst is so efficient. Theoretical modeling (#compchem) showed that gold nanoparticles activate the oxide surface toward the ethanol oxidation reaction. In other words, ethanol undergoes both partial oxidation and deprotonation immediately upon adsorption on the catalyst. Hence, our catalyst optimally prepares ethanol to the electrochemical oxidation process.
This knowledge, combined with the proposed fabrication route, may guide the development of electrocatalysts based on earth-abundant metal-oxides for ethanol valorization in Direct Ethanol Fuel Cells and for (photo)electrochemical water splitting.
Personally, I enjoyed very much this work, because metal-metal oxides interfaces are particularly challenging to deal with by #compchem. Also, I like very much to interact with my experimental colleagues and friends: they always have interesting problems, and collaborating together to find a solution is often the best part of the work. Very happy that computational modeling may help to understand the complex behaviour of these intriguing materials!
We presented this work at the fabulous #RSCPoster conference 2021. Here’s a pdf copy of our poster.
Chemical warfare agents put at stake human life and global safety. These compounds are extremely toxic, and their efficient detection is crucial.
By combining experiments and theory, we realized a new sensor, based on manganese oxide and gold nanoparticles, which has ultra-low detection limits and impressive selectivity towards an important simulant of the vesicant nitrogen mustard gas.
The manganese oxide nanomaterials were synthesized by chemical vapor-deposition (CVD), starting from a Mn(II) molecular complex. This compound can be easily vaporized, and gives manganese-oxide materials of high purity. Then, the manganese oxide surface was partially covered by gold nanoparticles.
Scheme of the synthesis of the Au-decorated manganese oxide nanomaterials
The material was then tested in the detection of a nitrogen mustard gas simulant (named dipropylene glycol monomethyl ether, DPGME). The results were exciting: the sensor showed high efficiency and selectivity, with a detection limit of 0.6 ppb.
All this work was done by our awesome experimental colleagues. However, I want to show you that also theoretical modeling (#compchem) has done its part here, by answering the question: how does this system work?
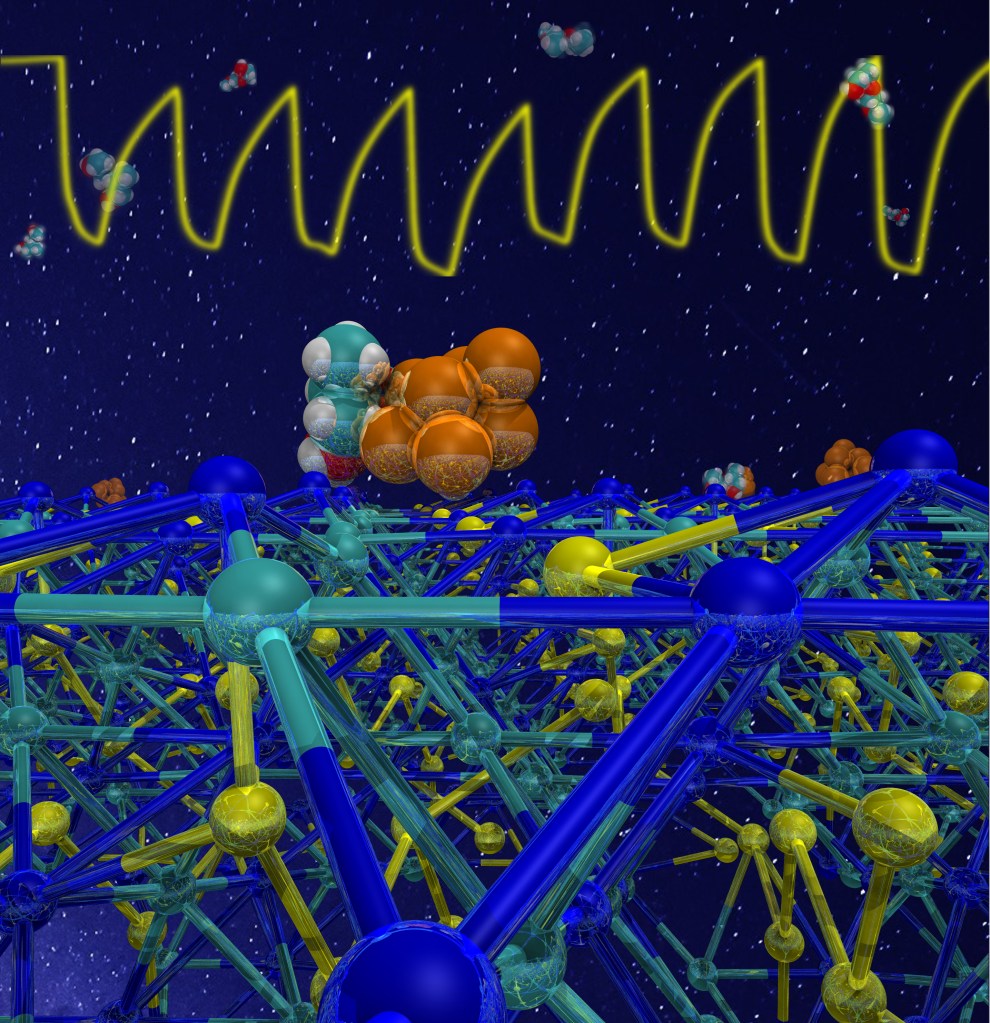
At molecular level, the sensing action depends on the contact of the analyte molecules with the active part of the sensor – the gold/manganese oxide layer. By using a density functional approach, we have seen that the molecule strongly binds to both gold and metal oxide, as shown in the picture below.
The mustard gas simulant DPGME (cyan,white,and red spheres) binds to both the Mn3O4, surface (ball-and-sticks) and gold (orange spheres). The top part of the figure shows the response of the chemoresistive sensor to the adsorbed DPGME molecules.
To check if this “molecular recognition” ability of the sensor was specific only to the target molecule, we modeled also the contact of ethanol with the sensor.
We found that while the mustard gas simulant is in intimate contact with both gold and Mn3O4, ethanol interacts only with the oxide surface, but not with gold. This can explain the higher response and selectivity of DPGME with respect to ethanol.
In short, the new materials have a low fabrication cost and remarkable sensing capabilities. The reason of their impressive performances in the detection of the mustard gas simulant is that the target molecule is anchored to both manganese oxide and gold.
I was really proud to present this work at the #RSCPoster2020 event – and I thank the organizers for this fantastic (and fun) opportunity.
By the way, if you still don’t know what a #RSCPoster conference is, follow #RSCPoster on Twitter, have a look to the many many great posters presented this year, and consider taking part to the 2021 edition!
Here’s our little contribution to this wonderful global event
Our poster presented at the 2020 #RSCPosterTwitter Conference
If you’d like more information this work – including the technical details of both modeling and experiments – these can be found in our published paper (see here for a free green open access version).
The best superpower ever? Being invisible, of course. How nice it would be to wear a magic cloak and escape boring meetings. Too bad that these tricks work only for ghosts, magicians, and superheroes. But in the atomic world, things are different. In this realm, our superhero is the smallest atom of the simplest organic acid: the proton of the formic acid molecule.
Many green chemistry applications involve organic acids and titania. So, the reactivity of formic acid on this material has been much explored, but its acid proton has escaped the most accurate experiments so far. Where is the missing proton?
By using computational methods #compchem, we’ve seen that the proton is shared between one oxygen of the formic acid molecule and one oxygen of the titania surface. At very low temperature, sharing is governed by real superpowers quantum effects, while, at room temperature, the proton moves very fast between the molecule and the surface – to see it in action, look at the movie! Anyway, in both cases, “sharing is caring”: it makes the acid proton “invisible” to experiments and protected from the attacks by bases.
The surface of titania acts like a protecting group for the formic acid proton. How does this work? Formic acid shares its proton with the surface, so it’s very difficult for other molecules to take it away! This protecting action of the surface could explain, for example, why carboxylic acids on titania, upon addition of amines, give high-value products (amides) at low costs for the environment.
Discovering the fate of the missing proton was a joy for my #compchem-ist’s eyes. Perhaps, it might become also useful for applications: indeed, it is just the acid proton that makes carboxylic acids on titania so interesting. Next step would be to study this process on different surfaces of titanium dioxide. This could help to understand how defects affect the reactivity of the acid proton.
On a personal note, my feeling is that our work has unveiled just a small part of the wonderful things that molecules can do on surfaces. But it’s so nice to find, from time to time, a little pearl in a shell.
Thanks @andrea_stangoni for designing such a marvelous artwork, thanks @angew_chem for this great honor, and for your lovely “sharing is caring” pun. We’re grateful to @ChemRXiv for hosting our preprint, all the people giving feedback on it, and the reviewers of the paper. No external funders to thank, this time. Instead, we send love to our old #compchem machines – the resources of a tiny group from a little institution planet in the Galactic Empire’s periphery.
The pearl in a shell also reminds me of a mollusc without shell. Along its 4y+ life, our project has faced abundant failures and rejections – yes, the familiar “been done already, nothing new, not worth financing” story, plus “simulations are, at best, an useful add“. No protective shell, and no Harry Potter’s cloak in those days. Yet, all those failures, critical comments, and rejections – even the most painful ones – prompted us to sit back and ponder previoulsy unseen aspects of the problem – and then, to work harder and deepen the analysis. This improved our work considerably. Also, posting a preprint on ChemRxiv gave us strong moral support during the final stages of the work.
We’ve all been there – things don’t always go as planned, and often let us down. But muggle tricks like persistence, faith, and time to cleanse mind and body spent on Twitter help a lot. My message? Keep calm and don’t give up: as someone else has said, “Hard times come, hard times go, yeah just to come again“.
Water. Always difficult to write something original about it, but let’s spend again a few words in celebration of this molecule. Water is present, or can be inserted in many porous hosts, like zeolites or MOFs. Not all of them love water. This time the question was: what does water do inside channels of similar size but different hydrophilicity?
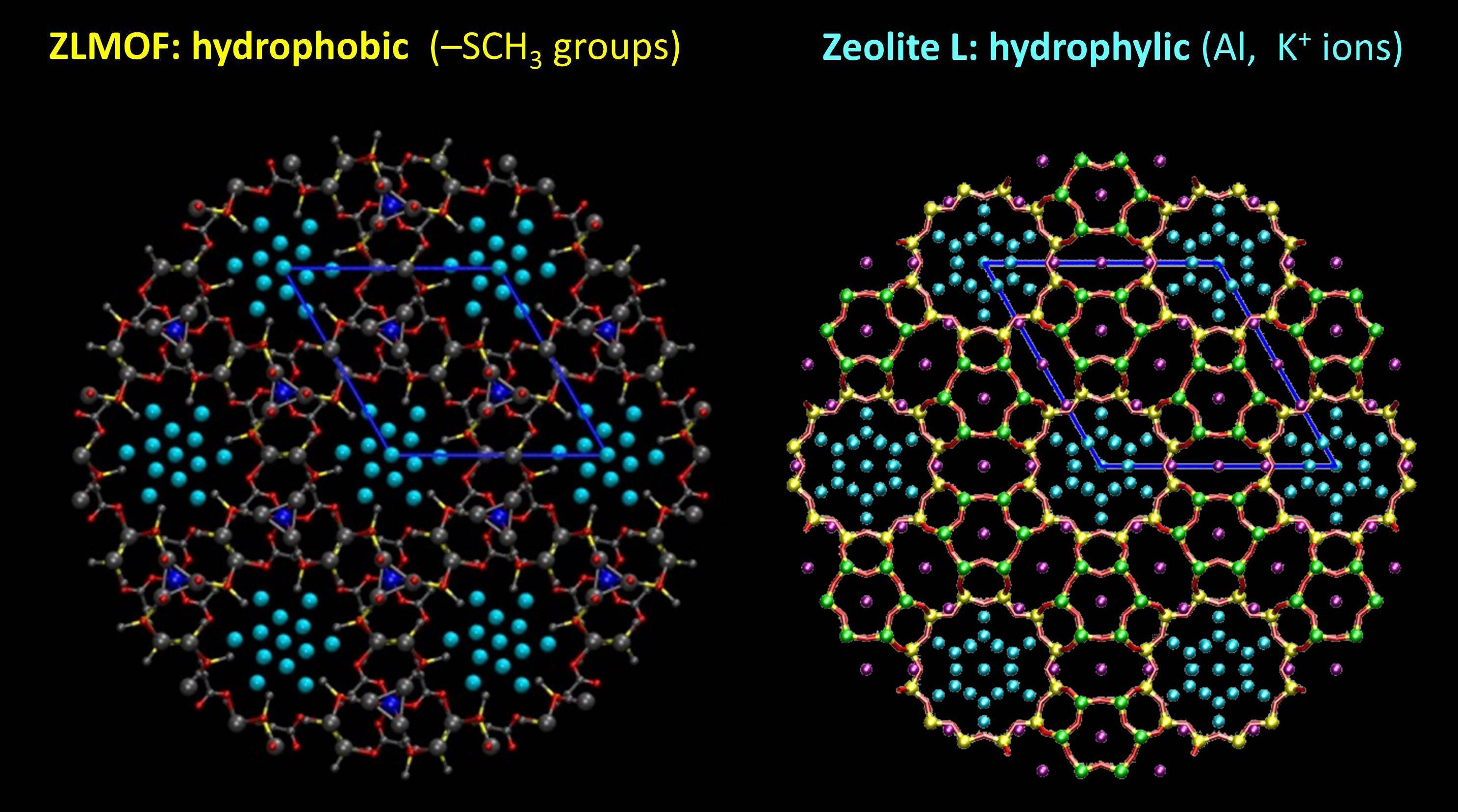
We modelled the behaviour of water in two porous materials. The first one is zeolite L, which is hydrophilic. The second one is a metal organic framework, or MOF, which has pores of similar size, but less affinity to water. Our starting point was the X-ray structure of the two materials, shown below.
X-ray structure of ZL-MOF and zeolite L, viewed perpendicular to the channel axis. In both materials, the water positions (cyan spheres) are partially occupied
The water distribution inside the pores looks very nice and symmetric. Unfortunately, the water positions are only partially occupied. So, by using the experimental water content as input, we optimized the structure of these materials, and here’s what we got.
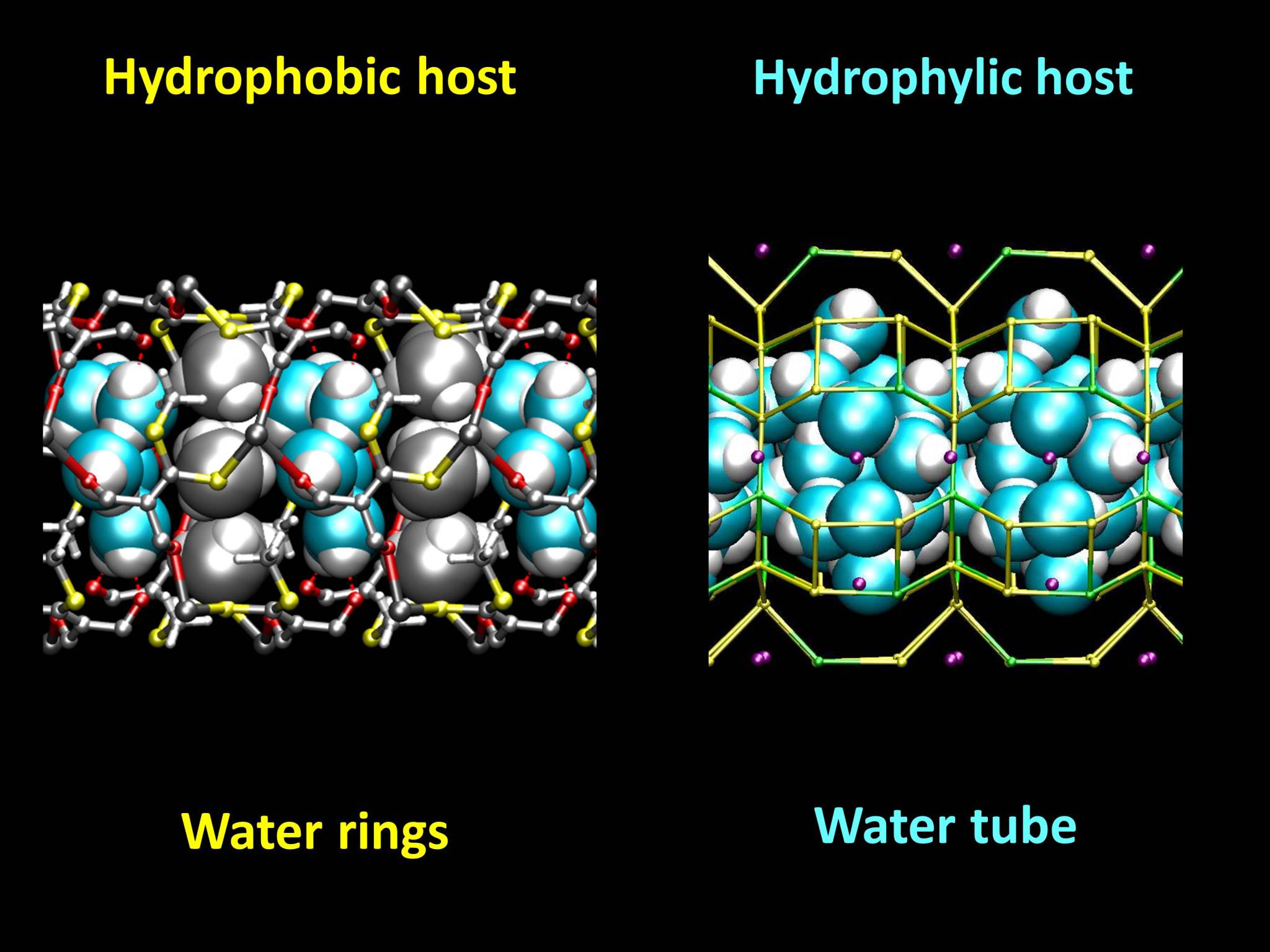
Optimized structures of ZL-MOF (left) and zeolite L (right), viewed along the channel axis. The hydrophobic methyl groups of the MOF (in gray) protrude inside the channel and force the water molecules to arrange in well-separated rings. In the hydrophilic channels of zeolite L, a continous water structure is formed.
We found that water stabilizes both materials, and that the shape of the water clusters inside the channels depends on the affinity of the hosts to water.
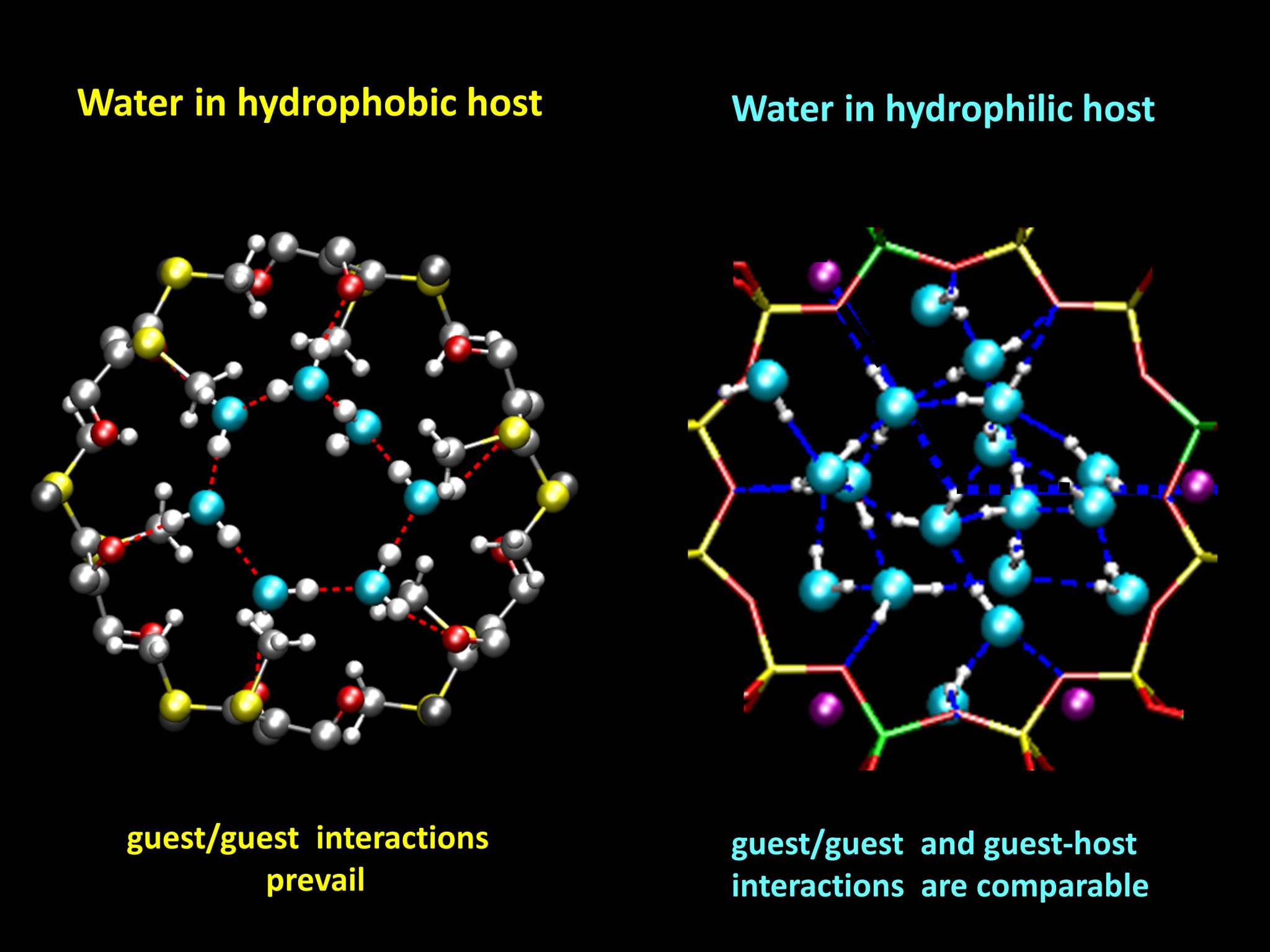
While the hydrophobic host contains water rings, kept together by water-water hydrogen bonds, the hydrophilic host contains a continuous water tube, stabilized by interactions with the zeolite and also by hydrogen bonds.
Optimized structures of ZL-MOF (left) and zeolite L (right) (front view). While the water rings in the MOF are dominated by water-water hydrogen bonds, the water molecules in zeolite L can interact very strongly also with the potassium cations (purple spheres) and the framework.
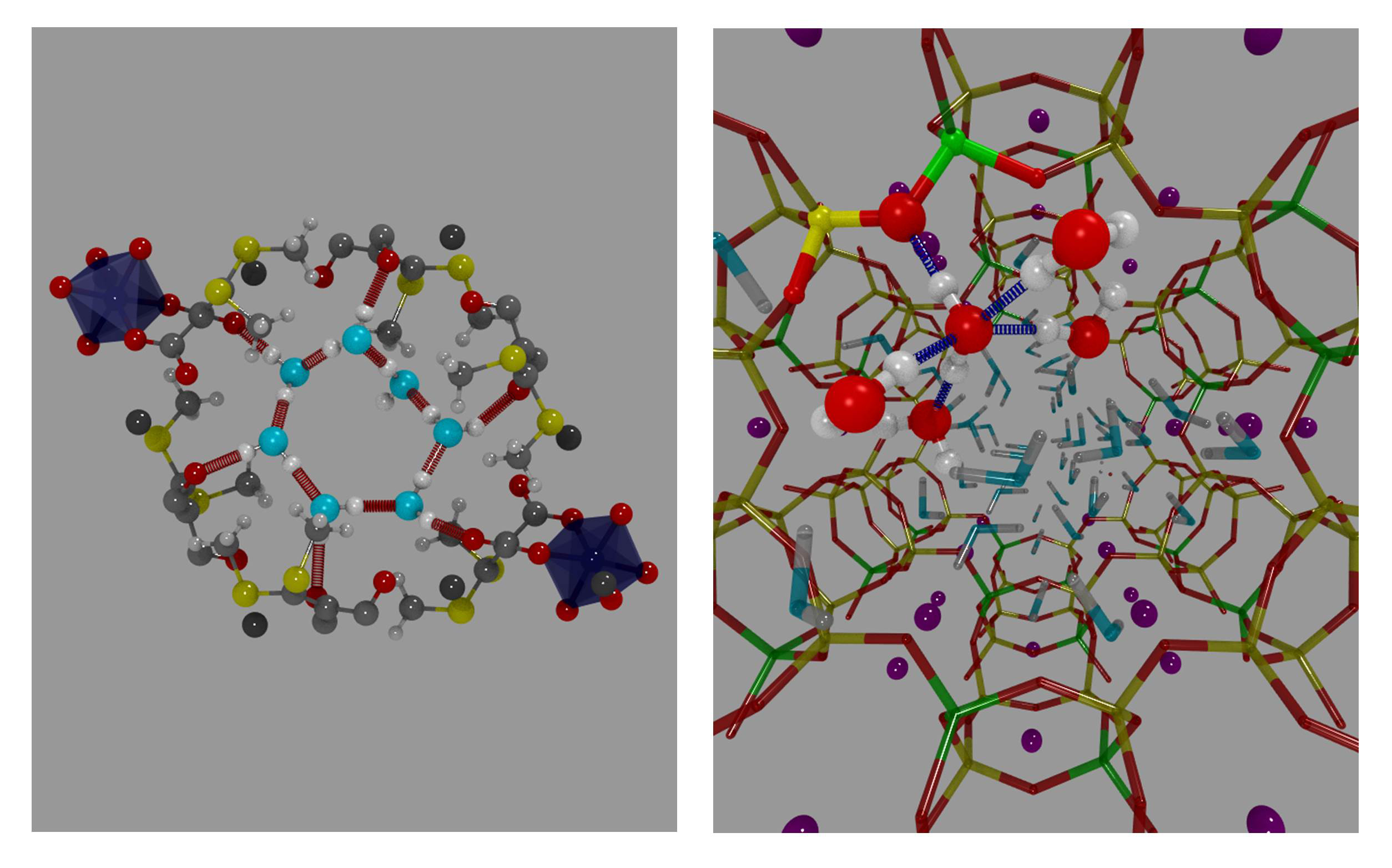
In the zeolite channels, some water molecules are surrounded by five strong hydrogen bonds. This structure is similar to water pre-dissociation complexes found in liquid water, and it might probably explain the high proton activity found in zeolite L.
Water rings in ZL-MOF (left), and water pre-dissociation complex (in red) in zeolite L (right)
Zeolite L is a promising material for solar cell applications, but the high proton activity inside the channels might damage some of the organic dyes that are incorporated as guests. Now we have identified a possible cause of the problem, and this might be a first step to improve the performances of these materials. Also, we hope that the atomistic insight on the water rings inside the MOF could help to exploit this material as host matrix for new compounds.
Personally, I much enjoyed doing this work: there’s always something to learn about confined water! The “driving force” for starting this work was an invitation, so many thanks to Michael Fischer and Robert Bell for organizing the Special Issue“Modelling Crystalline Microporous Materials” in the Zeitschrift für Kristallographie. If you like porous materials and #compchem, please have a look at this issue, it has many beautiful contributions. Also, thanks to ChemRxiv for hosting our preprint, and to the 2019 Twitter #RSCPoster conference, where this work was first presented as poster.
As much as you enjoy celebrating chemistry, doing research and running experiments, it comes a moment when you feel tired, disappointed, or sad. Sure, we all have those moments – and when they come, the warmth of the community might do a great deal (trust me – it works)!
Although the #RealTimeChem hashtag “operates 24/7“, there’s a special time of the year: the #RealTimeChem Week, aimed at raising awareness of the community and encouraging chemists of any level and discipline to join in. By following #RealTimeChem, you see lots of cool chemistry, and the fun is doubled if you also take part. It involves a “competition” with different award categories (sponsored by C&EN ), and other fun activities, like a creative chemistry cooking contest (sponsored by @ChemPubSoc_Euro), or a blog carnival. This year I am particularly excited, because i happened to be among the winners (thanks C&EN!) and this made my day.
Nonetheless, taking part and being inspired by all the great chemistry outside there, was equally exciting – both fun and instructive. For a taste of the awesomeness of the event, please take a look to this beautiful article from C&EN (which also contains fab pics from the winners/runners-up of all categories), or to Doctor Galactic’s blog , where you may find “delicious” images from the winners of the #Whatscooking? contest!
This is the right occasion to celebrate #RealTimeChem for bringing together people actively doing (or curious about) chemistry, its inventor Dr. Galactic, all the organizations supporting the amazing activities, and everyone that post their fun/inspiring/unexpected/awesome contributions to #RealTimeChem.
This year the theme of the week (as suggested by @g_laudadio and chosen by the community) was #Chem4Life – the chemistry of every day life. Being a computational chemist, I find it often hard to share my “everyday chemistry” with lab colleagues, especially those working with colourful transition metal complexes, or beautifully fluorescent stuff. Of course, nice molecular graphics or three-dimensional orbital plots nowadays help a lot to raise interest in theoretical/computational chemistry (#compchem), but what happens behind the scenes? What actually is “everyday #compchem”? The pictures I shared on Twitter were an attempt to capture such an aspect in a visual, immediate way! Here is it:
Wavefunctions in minimalist look!
As regards the #compchem hasthag, it has been around for long time. It is used among #compchem -ists to exchange research news about #compchem papers or codes, but also as a discussion forum, to ask for advice, to advertise a PhD or postdoc position… and so on. There is also a “community journal” named Computational Chemistry Daily, maintained by @janhjensen, to which everyone can contribute by tweeting a link to a paper (or to a code, blog post, graphics..) using the hashtag #compchem. It is extremely useful for keeping up with the field: I see it as a sort of community service, and love it. I’ve learnt a lot by following it, and met awesome people! It’s important for the computational/theoretical chemists of all ages, levels, and flavours of #compchem to have an informal place where they could meet and discuss. And I am grateful that this place exists… thank you so much all of you awesome #compchem-ists!
Overwhelmed with the increasing flow of new scientific discoveries and related literature? You’re not alone. We live in the information overload era: too much to read, too little time, and life is short. Probably we’d need more readable, shorter papers too. Why writing a long one? Perhaps, it might connect disciplines which speak different languages but have much in common. Like material science and mineral science.
Let’s start from the first one.
You can make materials for solar cells, optical devices or medical sensors by trapping molecules or nanoparticles inside a “host”. Once there, molecules are no longer free to move, like in a gas or a liquid. This process, called “confinement”, brings to life new properties, which were not present in the individual molecules and are very useful in technology. Energy transfer or information storage, for instance, are made possible by the organization of the confined molecules.
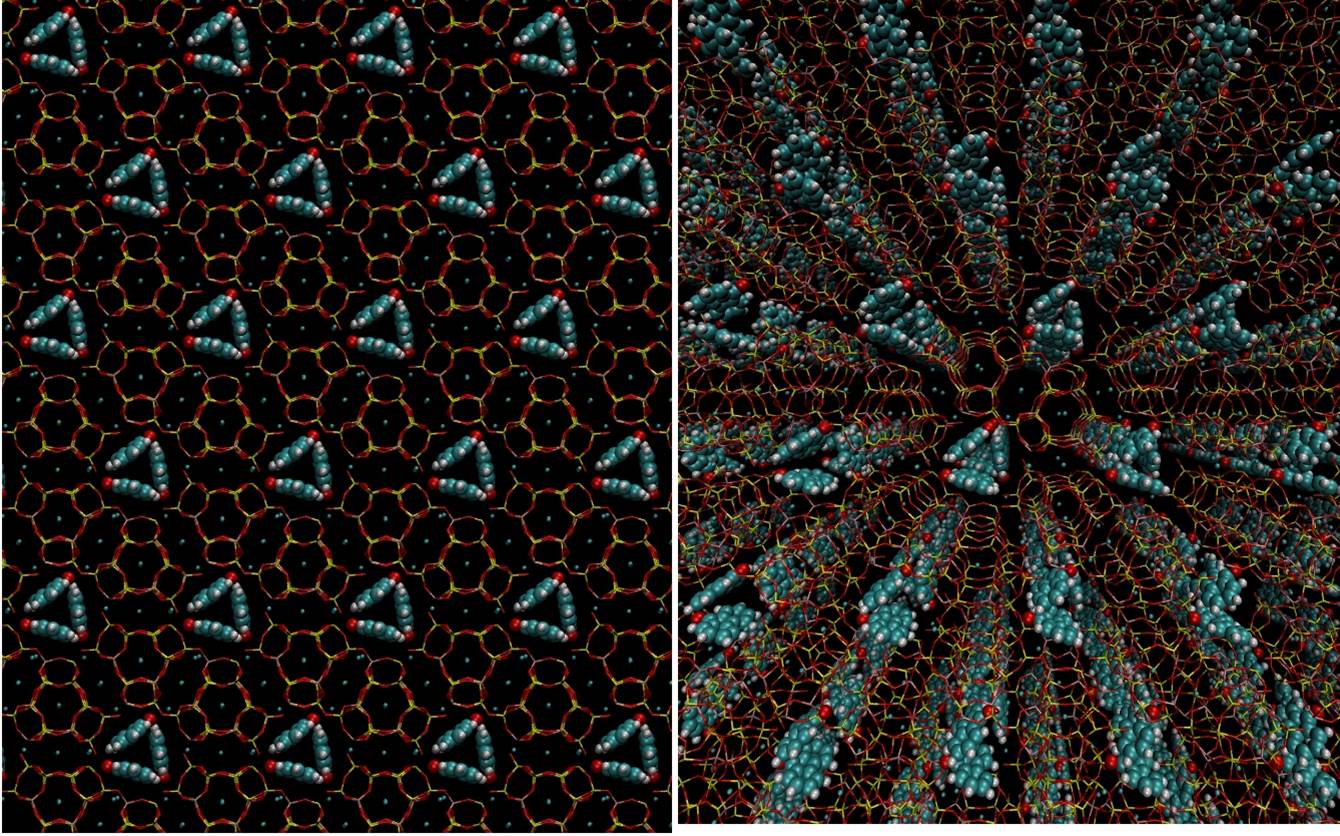
The regular cavities of zeolites do a great job in organizing guest molecules
Tiny smart objects such as molecular machines, motors and diodes, make good use of self-organization processes, which create order from apparent disorder by exploiting interactions between molecules. This task gets easier when molecules are confined in regular pores. Think of a buzzing swarm of bees, first frantically hovering in the air, and then accommodated in a honeycomb.
Similar to honeycombs, regular patterns of pores like those in zeolites can orderly accomodate small molecules or clusters. But if you want to entrap, say, enzymes, peptides, or large nanoparticles, you must use materials with larger pores. Some porous silicas have large honeycomb channels, while the cavities of metal organic frameworks display an amazing variety of size and shape. With those nice architectures awaiting to be filled, ordering molecules might appear like an easy task.
As you imagine, things are more complex. Perfect order cannot be achieved. All cavities would need to be uniformly occupied by the guests. This is going to be very unlikely, because molecules move a lot even when they’re confined… like bees in a hive.
Molecules in nanocavities are sort of like bees in a honeycomb: they form an organized colony (Artwork: Andrea Stangoni)
About bees, I had direct experience… as a child, I used to observe my dad opening up his hives to inspect them. This gave me the chance to “study” the behaviour of these awesome creatures inside their honeycomb.
Bees do not occupy all hexagonal holes in the frame, and move continuously around, without any apparent pattern. Hence they’re not perfectly ordered. In spite of this, the colony is amazingly organized, and performs an impressive number of complex tasks…. not just honey production!
Similarly, guest molecules confined in porous cages are not rigorously ordered. Yet they are organized, and the resulting host-guest materials can perform useful functions, which were absent in the free molecules. They can, for example, absorb and transfer photons like the antenna systems of plants and bacteria.
Now, the question is: can we improve the organization of the molecules and the performances of the materials? Well, first we should know how the molecules occupy the cavities, their orientation, spacing and so on. Are the guests aligned? Are they attached to the pore walls? What happens if water enters the pores? To find those answers, you should use several different techniques: each experiment will give you some pieces to compose the puzzle. And yes, computational chemistry helps a lot to figure our what happens inside the pores. Yet this remains a very difficult problem.
This is where mineral science might help.
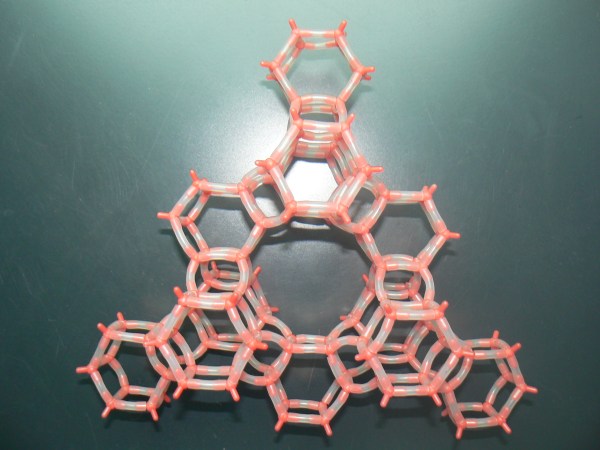
Regular patterns of cages are very common in the mineral world. Not long ago, for example, geologists found in Antartica a mineral with the same structure of zeolite Z-SM5, a well-known and widely used artificial industrial catalyst. That was indeed a big surprise! Natural zeolites are indeed amazing: their pores contain impressively stable structures formed by small molecules and cations. Just look at this water wire:
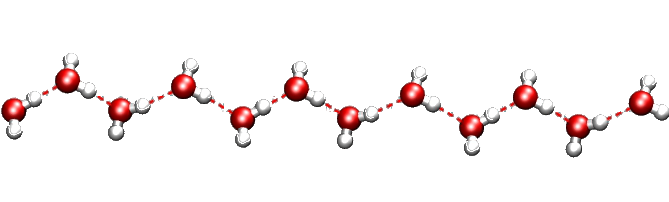
Water wire found in the channels of a natural zeolite
Contrary to what you’d expect, this chain is incredibly resistant to heat and pressure. First found in a rare mineral, it was named “one-dimensional ice”. But actually, our water wire “melts” at about 340 C inside the mineral framework! This is a great example of organized structure made by Nature. You can find many others: the most famous ones are perhaps gas hydrates. Several silica minerals have hydrate structures, which are also very common in man-made porous materials. Indeed, we should pay more attention to the close links between natural and artificial host-guest materials.
Natural porous minerals, the intriguing organization of their guests, and their response to mechanical stress can be an awesome source of inspiration in the quest of more robust and efficient materials. High pressure experiments with zeolites (and also some MOF’s) have already brought us new organized materials, along with many curious facts. But there’s so much yet to be discovered.
Perhaps, the problem with us (me included) and with our scientific era is that we don’t take enough time to relate with other disciplines. I’ve been so lucky to work with many awesome colleagues from the mineral, chemical and material science communities over the years, and it’s thanks to them that I wrote this review. One thing I learnt is that we should always try building bridges and strenghtening links between different fields because there’s nothing to lose, all to gain from a deeper exchange of ideas.
The title of this post is the literal translation of a proverb. The proverb means that Devil’s pot of wickedness sooner or later will boil – and, as there’s no lid, someone will see its content and reveal the truth. That’s the old innocent idea that, finally, justice will prevail over evil… well, I like it so much I use it as title. Rather than devils, this post is actually about pots and lids – of molecular size, of course.
As that’s not a Masterchef contest at the nanoscale, let’s get rid of the pot for the moment, and call it ‘container’. In the nanoworld there are many such containers, which can be filled with molecules. In this way, you can produce new materials with applications in various areas of technology: from solar energy to sustainability and human health.
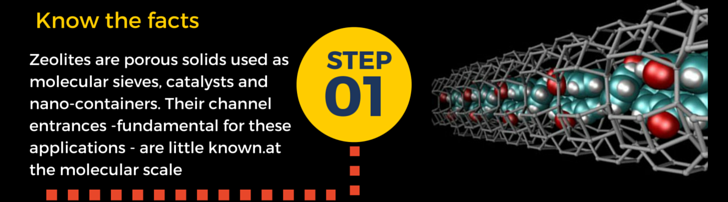
Our containers are named zeolites – porous materials which are commonly used as adsorbents and catalysts in various commercial, industrial, and even medical applications as well as in our everyday life. Also, if you fill zeolites with dye molecules, you’ll get materialsable to capture and transfer solar energy very efficiently. You would do it much easier if you first know how their pores look like.
In particular, how do their entrances appear to an incoming molecule? This question is our “step one”, because this information is really hard to get from experiments.
Fortunately, modeling comes to the rescue…. and that’s one of the reasons why I love so much doing #compchem (computational chemistry)!!
Step 2 revealed that the channel openings expose hydroxyl groups, and look somewhat like this:
Entrance of zeolite L channel, showing the terminal -OH groups and the channel accessibility.
Those terminal hydroxils can be condensed with other molecules, carrying specific groups, hence new properties and functionalities. Among them, the possibility of “closing” the pores. Why is it so important?
Zeolites are resistant to heat and pressure, and act as a protective shield around the dye. But every “pot” needs a “lid”: plugging the zeolite pore entrances, so that the dyes, once included, cannot escape into the environment, would further enhance their stability. This has already been done experimentally, by attaching at the channel entrances peculiar molecules nicknamed “stopcocks”. They consist of two “parts”:
the “tail”, which can penetrate zeolite pores;
the “head”, which is too big to enter the pore and remains outside, thus blocking (at least partially) the channel opening.
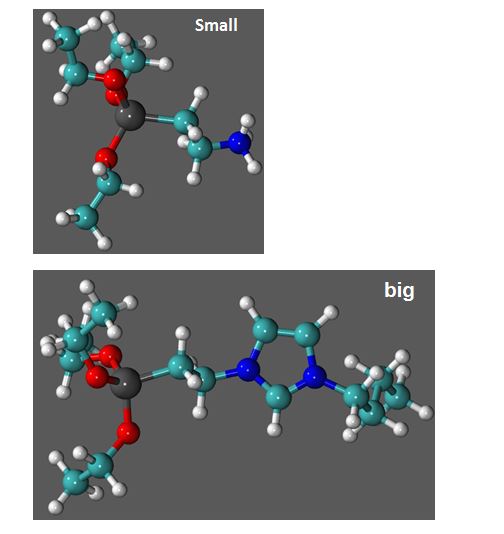
Two typical stopcocks, one with a small tail, and the other with a long, bulkier tail, are shown below.
Such “molecular stoppers” do indeed a great job in preventing molecules to escape from zeolites. However, there were no clear ideas about how these stoppers were attached to the pore entrance, and how much space they occupied. This knowledge would help finding better “lids” for our zeolite “pots”. How do we get it? Of course by modeling, as sketched in step 3 and 4.
Here’s what we learned:
stopper molecules prefer to bind aluminum sites at the channel entrance;
the tail group always penetrates inside the pore, while the head stays outside;
the extent of blocking depends on the stopcock.
In particular:
– small-tailed stopcocks are like partially opened “lids” : no full closure – bulky-tailed stopcoks behave like “corks”: full closure
So the zeolite pore may be fully sealed by one bulky stopper, like a molecular cork on a Prosecco nano-bottle. On the contrary, one “lid” (small stopper) leaves our “pot” partially opened. Fortunately, there’s enough room to attach a second small stopper to the opening, that can now fully be closed.
And this brings us to step 5…
… which could well be the end of this story, first told some time ago. Thank you for reading it!
Anyway, there’s an epilogue, which is perhaps the nicest part (“dulcis in fundo“). Using such information, obtained from modeling, experimental colleagues recently trapped indigo (that’s, your denim’s blue) in zeolite L, and blocked the channel entrances with two small stopcocks. In this way, they made a new pigment, exceptionally resistant, with an amazingly beautiful blue color. For me #compchemist, that blue was simply….. the color of happiness.
Requiring subscription: i) Indigo in the nanochannels of zeolite L (Woodtli et al, Dyes and Pigments, 2018, 149, 456); ii) Invention of the stopcock (Maas & Calzaferri, Angew Chem 2002, 41, 2284); iii) Official version of our article: Structure of Nanochannel Entrances in Stopcock‐Functionalized Zeolite LComposites,Angewande2015, 54, 11112
When we fill porous materials with dye molecules of the right size, we obtain useful compounds for solar energy technology. These compounds can transfer solar energy efficiently because pores and channels fit to the dyes “like a glove”. In this way, molecules are forced to stay in line, and energy can easily pass from a molecule to the next one in the line. If we knew in detail the structure of the dye arrays, we’d have better chances to improve these compounds.
Unfortunately, the precise positioning of the molecules inside the pores is very hard to determine. Recently, we solved this problem for a class of particularly efficient dyes filling the channels of zeolite L. Key to success was diversity within the team, which favored the combination of multiple techniques involving both experiments and calculations.
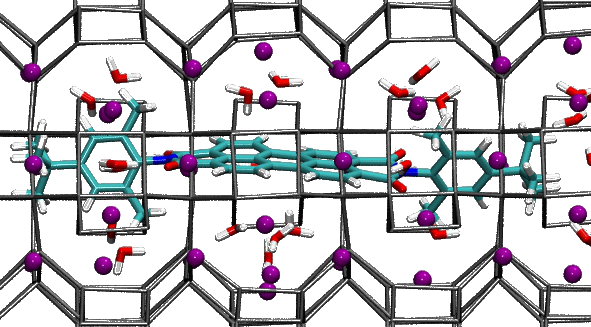
The useful properties of these materials arise from the arrangement of dye molecules inside the porous host, which depends on the interactions among molecules and with the porous host. After this work, now it seems we understand a little better these complex materials. Indeed, our dyes are linear, symmetric and fit to the zeolite channels. Yet they adopt a slightly asymmetric positioning to maximize the interactions with the zeolite cations, which stabilize the compound.
Perylene-bisimide dye (cyan) in zeolite L (gray). The purple spheres represent the zeolite potassium cations
This work also suggests some possible ideas to improve these compounds by modifying either the porous container (the “host”) or the dye molecule (the “guest”). In my view, this is also a good example of how computational modeling may help to rationalize experimental results in apparent contrast with each other, yielding a consistent picture of a useful and intriguing material.
Gigli et al. (2018) “Structure and Host–Guest Interactions of Perylene–Diimide Dyes in Zeolite L Nanochannels”J. Phys. Chem. C 122, 6, 3401-3418
TheTwitter Poster Conference is an annual event organized by the Royal Society of Chemistry, which consists of sharing chemical research using tweets. You may take part either by tweeting an image of your poster or by commenting on other poster (doing both is better, in my view). As in a traditional conference, you see great research, are asked interesting questions, meet old friends, and come in contact with new colleagues or potential collaborators. Only, at the twitter conference this happens 24 hours non-stop on global scale; so, it’s a good idea to get there prepared!
I enjoyed so much my 2017 participation that I couldn’t miss the 2018 edition. Of course, it was awesome, and I am grateful to the organizers, the sessions’ chairs, and all participants, particularly those with whom I interacted. Indeed, I have learnt new stuff, seen exciting science, been inspired, without moving from my office and paying any conference fees. Really cannot ask for more. Many thanks to all of you!
Tweeting posters is not trivial. For optimal readability, you should keep into account, for example, that mobile phones have small screens, and that Twitter images are resized and cropped down – so, it would be better to prepare the poster in landscape format. These and other useful tips can be found in this excellent post. I came across it when the event was over, but it would surely be useful in the future.
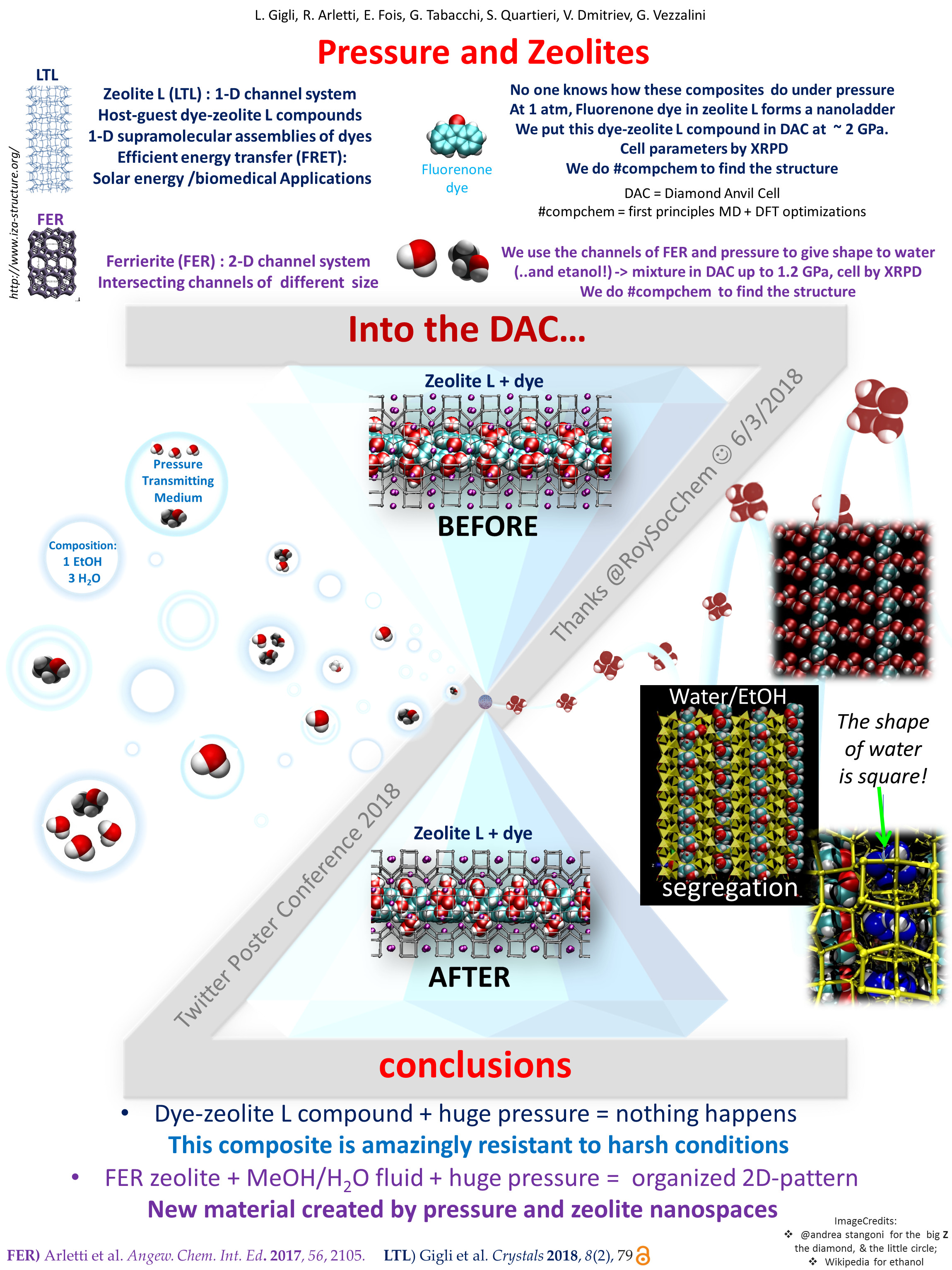
Below you can find my poster, illustrating the fruitful collaboration between calculations and diffraction experiments at high-pressure conditions.
Besides water and ethanol in ferrierite (discussed in this post), the poster shows our new work on a host-guest compound of zeolite L and fluorenone dye under high pressure. These host-guest materials have excellent optical properties, useful for many applications, from solar cells to sensing in medical technology. Knowing their structure and working principles could help improve their performances – that’s why we try so hard to understand dye-zeolite composites at molecular level.
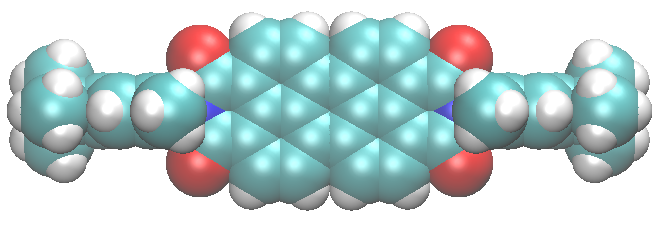
Basically fluorenone inside the channels of zeolite L forms a molecular ladder, which is very stable at room conditions because the carbonyl groups of the dye interact very strongly with the potassium cations of the zeolite.
Is this peculiar structure also stable under GPa pressures?
According to experiments and simulations, the answer is apparently yes! Our composite maintains its structure, and the interactions between the dye and the zeolite cations become stronger. The exceptional resilience of this material to compression highlights its outstanding mechanical properties. These are important to extend the application of dye-zeolite composites beyond room-pressure conditions.









 Discovering the fate of the missing proton was a joy for my #compchem-ist’s eyes. Perhaps, it might become also useful for applications: indeed, it is just the acid proton that makes carboxylic acids on titania so interesting. Next step would be to study this process on different surfaces of titanium dioxide. This could help to understand how defects affect the reactivity of the acid proton.
Discovering the fate of the missing proton was a joy for my #compchem-ist’s eyes. Perhaps, it might become also useful for applications: indeed, it is just the acid proton that makes carboxylic acids on titania so interesting. Next step would be to study this process on different surfaces of titanium dioxide. This could help to understand how defects affect the reactivity of the acid proton.