

When we fill porous materials with dye molecules of the right size, we obtain useful compounds for solar energy technology. These compounds can transfer solar energy efficiently because pores and channels fit to the dyes “like a glove”. In this way, molecules are forced to stay in line, and energy can easily pass from a molecule to the next one in the line. If we knew in detail the structure of the dye arrays, we’d have better chances to improve these compounds.
Unfortunately, the precise positioning of the molecules inside the pores is very hard to determine. Recently, we solved this problem for a class of particularly efficient dyes filling the channels of zeolite L. Key to success was diversity within the team, which favored the combination of multiple techniques involving both experiments and calculations.
The useful properties of these materials arise from the arrangement of dye molecules inside the porous host, which depends on the interactions among molecules and with the porous host. After this work, now it seems we understand a little better these complex materials. Indeed, our dyes are linear, symmetric and fit to the zeolite channels. Yet they adopt a slightly asymmetric positioning to maximize the interactions with the zeolite cations, which stabilize the compound.

This work also suggests some possible ideas to improve these compounds by modifying either the porous container (the “host”) or the dye molecule (the “guest”). In my view, this is also a good example of how computational modeling may help to rationalize experimental results in apparent contrast with each other, yielding a consistent picture of a useful and intriguing material.
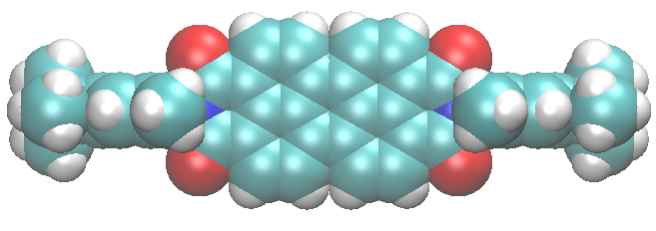
Gigli et al. (2018) “Structure and Host–Guest Interactions of Perylene–Diimide Dyes in Zeolite L Nanochannels” J. Phys. Chem. C 122, 6, 3401-3418

One thought on “Understanding an efficient light harvesting material”