When we fill porous materials with dye molecules of the right size, we obtain useful compounds for solar energy technology. These compounds can transfer solar energy efficiently because pores and channels fit to the dyes “like a glove”. In this way, molecules are forced to stay in line, and energy can easily pass from a molecule to the next one in the line. If we knew in detail the structure of the dye arrays, we’d have better chances to improve these compounds.
Unfortunately, the precise positioning of the molecules inside the pores is very hard to determine. Recently, we solved this problem for a class of particularly efficient dyes filling the channels of zeolite L. Key to success was diversity within the team, which favored the combination of multiple techniques involving both experiments and calculations.
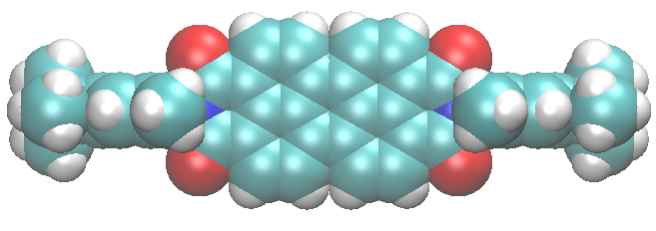
The useful properties of these materials arise from the arrangement of dye molecules inside the porous host, which depends on the interactions among molecules and with the porous host. After this work, now it seems we understand a little better these complex materials. Indeed, our dyes are linear, symmetric and fit to the zeolite channels. Yet they adopt a slightly asymmetric positioning to maximize the interactions with the zeolite cations, which stabilize the compound.
Perylene-bisimide dye (cyan) in zeolite L (gray). The purple spheres represent the zeolite potassium cations
This work also suggests some possible ideas to improve these compounds by modifying either the porous container (the “host”) or the dye molecule (the “guest”). In my view, this is also a good example of how computational modeling may help to rationalize experimental results in apparent contrast with each other, yielding a consistent picture of a useful and intriguing material.
Gigli et al. (2018) “Structure and Host–Guest Interactions of Perylene–Diimide Dyes in Zeolite L Nanochannels”J. Phys. Chem. C 122, 6, 3401-3418
How can a snake swallow a mouse bigger than its mouth?
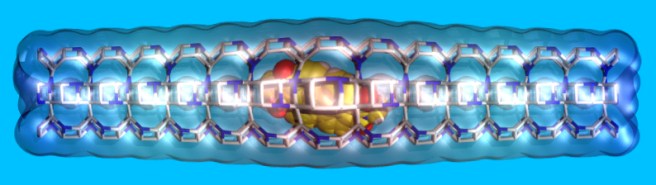
Weird as it seems, questions like this emerge very often at the molecular scale. For example, we can fill porous materials with molecules larger than the diameter of the pores: in this way, we may obtain devices for energy and health applications. What makes this useful process possible? Flexibility is the key: both the porous host (the “snake”) and the molecule (the “mouse”) must deform for the process to occur. But here, contrary to the mouse-snake case, cooperation between the two partners is needed.
We captured the passage of a bulky molecule through the very narrow opening of one of these pores. We did this by computer simulations, because it is very hard to get such information experimentally. To get an idea of what we found, you don’t even need to read the paper – and i’m not kidding. Just look at the movie below!
What we’ve seen first, is that the pore is slightly larger at its entrance. This surely helps the molecule to go in.
Second: contrary to the mouse, which would escape the snake as fast as it could, the molecule is indeed “magically” drawn to the pore entrance – by electrostatic forces.
“So what?” – you may say.
Keep in mind that the molecule is still larger than the pore opening. No kind of “fatal attraction” could do the trick, in a world of rigid bodies.
We’ve found that the molecule can pass through the opening and slip inside the pore only because it’s flexible, and its motion is “in tune” with the vibrations of the porous matrix. All this factors cope to make the entrance process more favorable than the exit process – that’s why the molecule gets finally swallowed by the pore, and remains trapped inside the material.
For me, it was very nice to see how bulky molecules manage to pass through narrow openings and travel inside a porous material. But finding out the reason why they stay inside was, probably, even more exciting: because it explains how materials of this kind can form and remain stable. Which is exactly one of the things you may need, in the quest of easier and smarter ways to produce better materials.
As we have to give credit where credit is due, i must confess that i borrowed the mouse-and-snake idea used in this post. But you’ll never know from whom. Me neither: (s)he was an anonymous referee of the paper. I am very grateful to this person: i can hardly imagine a nicest way to sketch our work.
Update:
Many thanks, of course, also to ChemComm for the cover!
What do a spacecraft, a breathalyzer, and carbon monoxide have in common? Nothing at all – you’d think. And you’d be wrong! All three give you information on things that you cannot directly see, touch or measure. A spacecraft can capture some signal and send you beautiful images of a planet. With the help of a breath tester, a policeman may deduce the alcohol content in your blood. And using carbon monoxide, researchers may find highly reactive centers on materials surfaces. Let’s focus on the latter and see how it works!
Left image: the Soyuz spacecraft (source: Wikimedia commons). Center image: a breathalyzer (source: photograpy by Elza Fiúza/ABr, distributed under a CC-BY 3.0 license). Right image: carbon monoxide (blue=carbon; red=oxygen).
When a molecule comes in contact with surface atoms, its properties change. By measuring these changes, you get information on the surface sites interacting with the molecule. Molecular vibrations – that you can measure by infrared spectra – provide very useful information: the vibration of carbon monoxide is very sensitive to the type of surface sites. That’s why this molecule is used to identify active centers on catalytic materials, such as titanium dioxide.
How does carbon monoxide (CO) bind to surface atoms? If you’re a chemistry student, you (should) know very well how CO interacts with molecules and ions. You’ve learned that this molecule can work both as a donor and as an acceptor of electron density. Well, what’s nice, is that this happens also on surfaces, and you can see it experimentally.
Let’s see this step-by-step. Carbon monoxide is a peculiar molecule. When it acts as a donor, charge flows to its bonding partner, which could be, for example, a metal cation. This process strengthens the C−O bond and increases its vibration frequency. This means that, in the infrared spectrum of the sample, you’ll find the CO band at higher frequencies – “blue-shifted” – with respect to the free, unperturbed molecule. But carbon monoxide can also accept electron density from its bonding partner. If this occurs, the C-O bond becomes weaker: its stretching frequency decreases, and you’ll see a “red-shifted” CO band in your spectrum.
Carbon monoxide is colorless, odorless, and highly toxic – a true and unmerciful silent killer. It binds to iron(II) in hemoglobin, and this prevents the delivery of oxygen to the human tissues. This is a – very unfortunate – case where carbon monoxide acts at the same time as a donor and as acceptor. The bond is synergic: CO donates to the metal, the metal back-donates to CO, and these two mechanisms reinforce each other:– that’s why it kills. This synergy occurs in many molecular complexes of transition metals and ions – often with less dangerous consequences. It’s less known on metal oxide surfaces, but it may happen as well. Is this the case of TiO2?
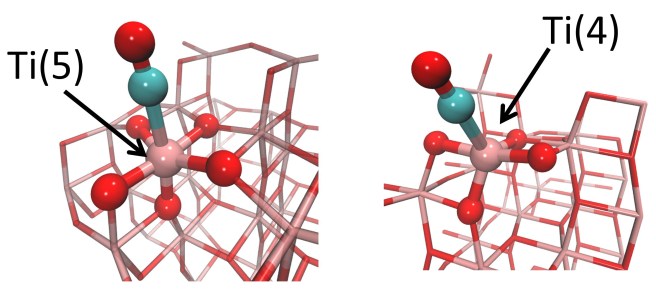
Not apparently, because carbon monoxide can only be a donor towards Ti cations – they are Lewis acids, and cannot give back electron density. The lower is their coordination number, the stronger is their acid power. For example, a Ti cation coordinated by 4 oxygens – Ti(4) – should be a stronger acid than one bound to 5 oxygens -Ti(5).
Researchers use carbon monoxide to explore the activity of surface cations and deduce their environment, in particular the number of oxygen neighbors. This information connects the reactivity of a catalytic center to its molecular structure, and may help them to improve the catalyst. Practically speaking, they send carbon monoxide on a TiO2 sample and measure the infrared spectrum. The rule is simple: the higher the frequency of the CO band, the more reactive are the Ti sites on the sample, and the lower their coordination number.
The image shows a 5-coordinated Ti center (left) and a 4-coordinated Ti site (right) on anatase-TiO2 surfaces
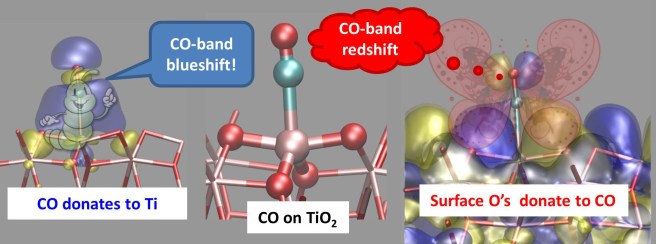
So if you had a TiO2 sample with Ti(5) sites, and a second one with Ti(4), what would you get from the experiment? “The second sample should show a more blue-shifted CO band, because Ti(4) is a stronger Lewis acid”. If you answered this, you’d be wrong… because we actually did the experiment, checked with calculations, and found the contrary. We found that CO on Ti(4) gives a less blue-shifted band – even if Ti(4) is a stronger Lewis acid. Just as if a breathalizer estimated a lower alcohol content in a drunker driver. This could happen only if a sort of magic potion neutralized the effects of alcohol (something similar exist in real life, but it’s a mineral and belongs to the large family of zeolites). Similarly, our carbon monoxide on Ti(4) should have received an antidote against the loss of electron density. The antidote could only be electron density: but where did it come from? Simply from the oxygen atoms bound to Ti(4): they are close enough to CO and ready to help.
In short, what happens is that CO donates electron density to Ti, but the surface oxygens donate electron density to CO. The first process strenghtens the C-O bond, but the latter has opposite effects. As a result, you find the CO signal at frequencies lower than expected. The two mechanisms are sketched in the figure below – my attempt to explain in a simple way the two-fold nature of the Ti-CO bond on titanium dioxide surfaces.
So, if you see high frequency bands in an infrared spectra of CO, please be warned: not necessarily they are due to very reactive sites on TiO2 surfaces. And also keep in mind that carbon monoxide gives you indirect information on your sample. Its signal can be influenced in complex ways by several factors – you might misinterpret your data, based on simple rules. From a practical viewpoint, i think that you should be aware of this, especially if you’re working on CO, or titanium dioxide materials. More speculatively, this story might help us to better understand how molecules interact with surface atoms. The complex, delicate balance of molecular-scale interactions is at the origin of technologically important phenomena – reactivity, catalysis, photocatalysis, just to mention some of them. Understanding these interactions more deeply could help us to improve their practical applications. Much effort is still needed, but it’s worth doing!
This research by our group has been published recently (Deiana et.al., ChemPhysChem2016, 17, 1956; 10.1002/cphc.201600284). It was also sketched in a short summary, and by an infographics in a previous post. Here i used other words to tell the same story, because i feel it’s important to make research results accessible to a larger community.
During this weekend i tackled a challenging task: to try to explain one of my recently published papers with an infographic. My first thought was to write a blog post (maybe i’ll do it as well), but i was intrigued by the idea of lumping a couple of years of work into a few tiny lines. Although attracted by the immediacy of infographics, i never used this tool, and it sounded just the right moment to give it a go. So, i went to the Canvas site and chose a fitness club advertisement as a template. After a bit of playin’ around, that’s what i’ve got:
I admit i’m quite happy with it, even if the making process was not plain sailing at all, at least for me. As a first-time user, i think that there’s room for improvement, and i’ll probably do some other attempts. Actually, i enjoyed creating this infographic!
I have published it (the infografic, i mean) in figshare (acceptance rate: 100%, publication fares: 0 €). That’s openaccess – free to download and use. Unfortunately, that’s not true for the paper – not enough funds to make it openaccess as well. Anyway, if you might want to give it a look, here’s the link:
Deiana, C., Fois, E., Martra, G., Narbey, S., Pellegrino, F. and Tabacchi, G. (2016), On the Simple Complexity of Carbon Monoxide on Oxide Surfaces: Facet-Specific Donation and Backdonation Effects Revealed on TiO2 Anatase Nanoparticles. ChemPhysChem. doi:10.1002/cphc.201600284
Another short explanation can be found here, with links to additional material.
To understand how a motor works you have to know how it is put together and how it can be disassembled. This is true at the molecular level as well. Amazingly complex molecular motors and machines are fabricated everyday, but how do they break down into their constituent pieces? Attracted by this question, we modeled two such species – called ‘rotaxanes’ – and made them break apart.
Typically, rotaxanes are made by two molecules: a ring-shaped one, the “wheel”, and an approximately linear one, the “axle”. What is great about them, is that you can modify the interactions between the ring and the axle by using an external signal. Which means that you can control the movements of these components through light, for example.
A ring (R)An axle (EE)Another axle (ZZ)
Using the three components shown above – one ring and two axles – you can build two different rotaxanes: R-EE, and R-ZZ. Both disassemble – or ‘dethread’ – when the axle exits from the ring. This process occurs differently for the two species: While the EE-one (video 1) dethreads in one single step:
the dethreading of the ZZ-axle (video 2) passes through an intermediate and a transition state:
Analyzing the two simulations, we identified the interactions between the molecular components which control the process at the molecular level. For example, we have seen that the elliptic shape of the ring opening is very important, as it can recognize the two different configurations of the terminal groups of the axles. This insight might help in the construction of light-powered molecular devices, of which these rotaxanes are important building blocks.
The results of this work are discussed in our paper (1), which is part of the ChemPhysChem special issue on molecular machines. You may get a nice picture of the field by taking a look at this issue. Its contributions well illustrate the concept that molecular machines -especially natural ones – are incredibly complex and fascinating systems, and we are just beginning to understand their inner workings.











{kind=link}